Summary
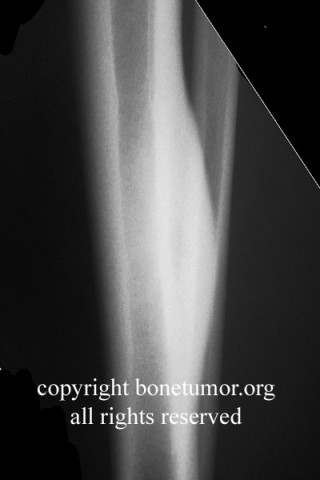
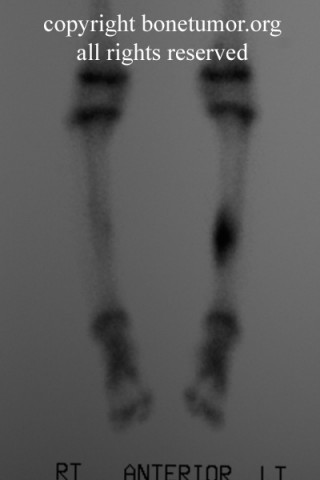
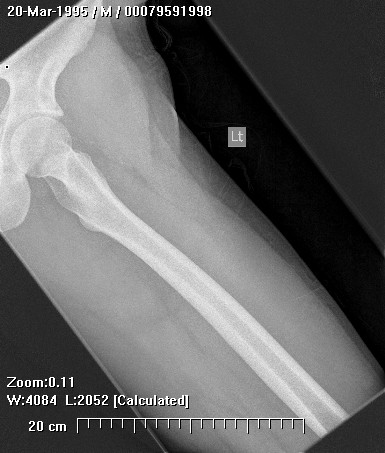
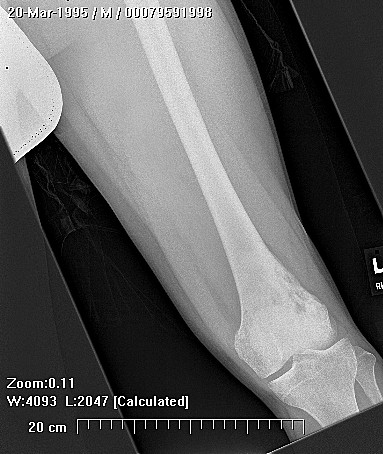
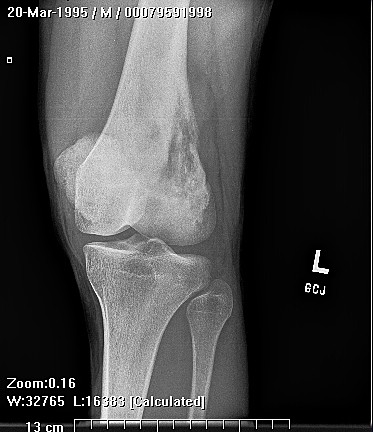
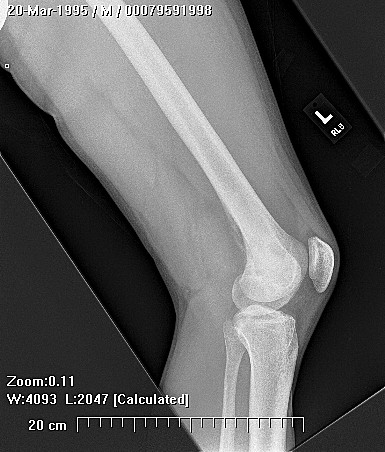
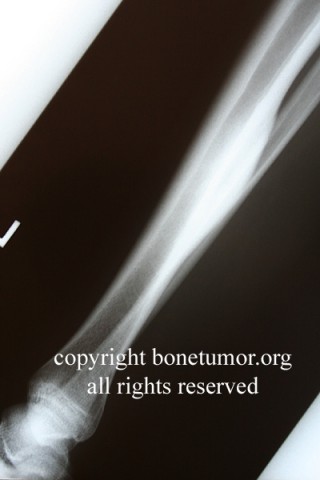
Erdheim–Chester disease (also known as Erdheim–Chester syndrome or polyostotic sclerosing histiocytosis) is a rare disease characterized by the abnormal multiplication of a specific type of white blood cells called histiocytes, or tissue macrophages (technically, this disease is termed a non-Langerhans-cell histiocytosis). Usually, onset is in middle age. The disease involves an infiltration of lipid-laden macrophages, multinucleated giant cells, an inflammatory infiltrate of lymphocytes and histiocytes in the bone marrow, and a generalized sclerosis of the long bones
Complete Information on this Tumor
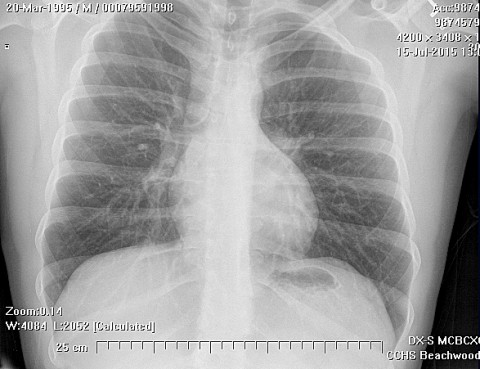
Erdheim-Chester disease is a rare systemic disorder, which is characterized by mononuclear infiltrate consisting of the lipid laden foamy histiocytes, which are found in various skeletal and extraskeletal organs. Most common sites involved are long bones,central nervous system, retroperitoneum, lung, heart, liver, spleen, skin and orbit. On histological examination there is infiltration of lipid laden histiocytosis within the connective tissues of the affected organ and positive with antibodies against CD68 on immunohistochemistry.
Patients mostly present with pain in lower extremity. The involvement is usually bilateral and symmetrical. There is specific involvement of the appendicular skeletal, though involvement of the axial skeleton has also been reported. There can be associated general symptoms like fever, myalgia, weight loss, night sweats and flu like symptoms. The patient with systemic involvement can present with neurological symptoms, diabetes insipidus, exophthalmos, skin xanthomas and low back pain due to retroperitoneal fibrosis.