Summary
HMOCE (hereditary multiple osteochondral exostosis) is a rare, autosomal dominant inherited disease where multiple osteochondral bone tumors occur on the surfaces of the bones. The severity of the disease can vary from mild to very disabling. A small number of the tumors may develop into low grade chondrosarcomas.
Complete Information on this Tumor
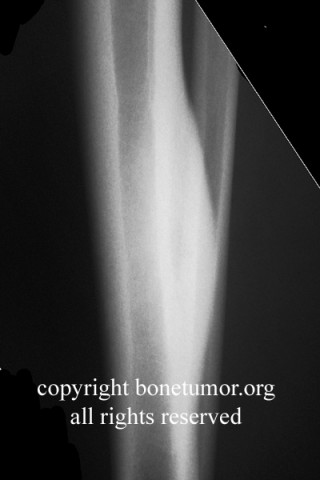
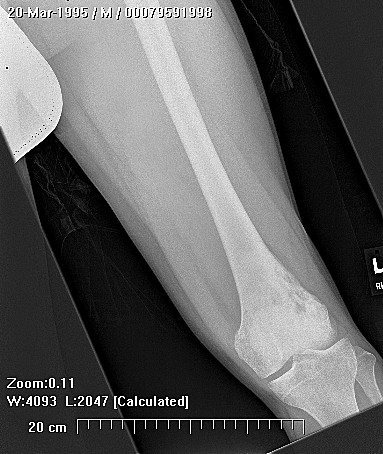
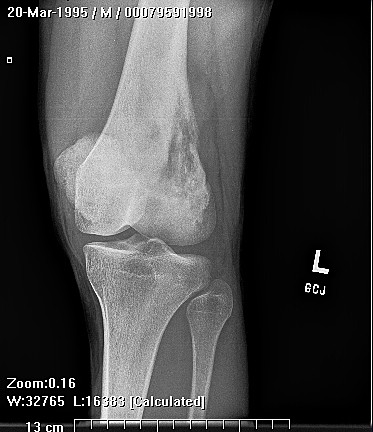
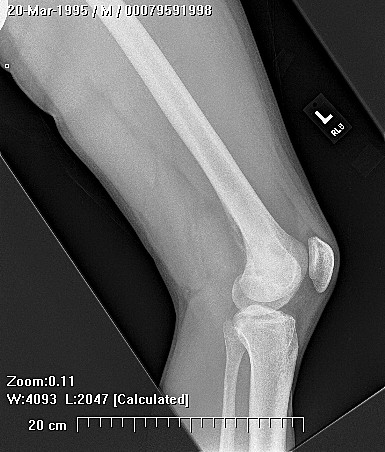
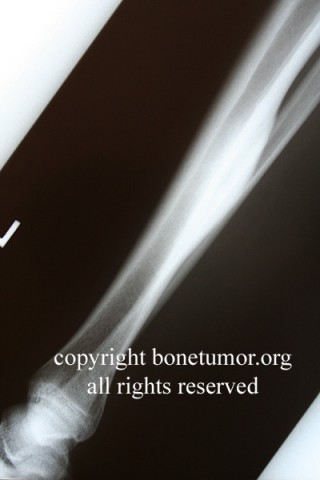
HMOCE is a rare, autosomal dominant condition where an genetic error of bone growth results in the formation of multiple osteochondromas. The condition can range from a mild nuisance to a very severe, disabling, and even potentially deadly problem depending on the number and location of the lesions.
As HMOCE is an autosomal dominant condition, it is genetically heterogeneous and it has been linked to three loci including 8q24.1 (EXT1), 11p11-12 (EXT2), and 19p (EXT3). EXT1 and EXT2 are proposed tumor supressor genes which may be lost in HMOCE, resulting in the growth of the lesions.
The risk of malignant transformation to chondrosarcoma in hereditary multiple osteochondromatosis is unknown, but may be 25-30% compared to approximately 1% for a solitary osteochondromas. The risk of malignant degeneration increases as the number and size of the osteochondromas increases. In general, a sessile lesion is more likely to degenerate into sarcoma than an exostosis.
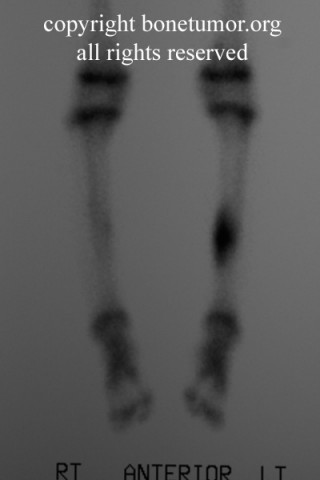
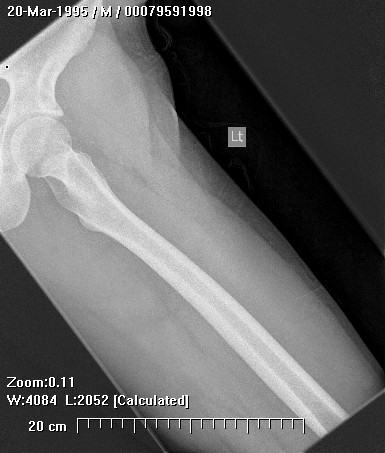
About 50% of affected children have identifiable lesions by age 5. Patients present with disfigurement or with pain induced by pressure on surrounding soft tissue structures. They may have short stature, limb-length discrepancies, valgus angulation of the knee and ankle, bowing of the radius with ulnar deviation of the wrist, and subluxation of the radiocapitellar joint. The ulna and fibula are more affected than the radius and the tibia. For this reason, differential growth of the paired bones it may lead to angular deformities of the forearm, wrist, leg, and ankle. Also, lesion clusters at the metaphyseal ends of the bones near the joint may lead to loss of motion, hip dysplasia, joint subluxation, nerve compression, vascular pseudo-aneurysm, cord compression, and pain.
Indications for surgical removal include persistent pain, functional impairment, joint subluxation, neuropraxia, and others. Early surgical corrections of deformities of the forearm has been advocated to improve function. However, the actual functional improvement has been questioned, and surgery may be safely delayed until after skeletal maturity. Significant angular deformities of the lower limb can be reduced or eliminated with re-alignment procedures. Surgical removal of troublesome lesions or lesions which appear to possess malignant potential is associated with a good outcome. Significant angular deformities of the lower limb can be reduced or eliminated with re-alignment procedures, which should improve function and reduce pain. Early surgical corrections of deformities of the forearm has been advocated to improve function. However, the actual functional improvement has been questioned, and surgery may be safely delayed until after skeletal maturity. Complications occur in approximately 10-12% of cases following elective excision of osteochondromas, including nerve injury, arterial laceration, and fracture. For this reason, the lesions should not be removed just because they exist. Indications for surgical removal include persistant pain, functional impairment, joint subluxation, neuropraxia, and others.
After surgery, patients with HMOCE form larger scars than usual, which is probably caused by the underlying HS synthesis defect in some way.
Wirganowicz, PZ Journal of Pediatric Orthopedics. 17(4):455-459, July/August 1997
Alman, BA. Multiple Hereditary Exostosis and Hedgehog Signaling: Implications for Novel Therapies JBJS -Am. 2009 :91 Suppl 4:63-7
Steiber and Dormans, J Am Acad Orthop Surg. 2005 Mar-Apr;13(2):110-20