Summary
Description
El sarcoma de Ewing es un tumor altamente maligno que afecta mayormente a niños(as). Comúnmente se encuentra en el fémur, tibia, humero y pelvis.
People and Age
Aunque esta enfermedad puede afectar a personas en varias edades, es más común entre aquellas de 1-20 años.
Symptoms and Presentation
La presentación clínica del sarcoma de Ewing incluye dolor e hinchazón que dura semanas o meses.
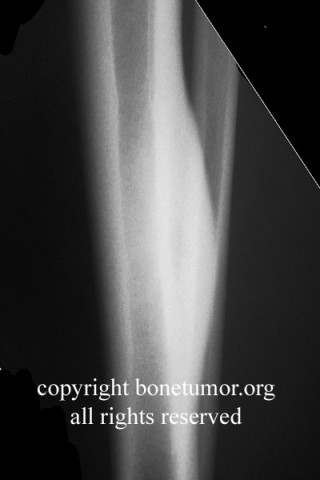
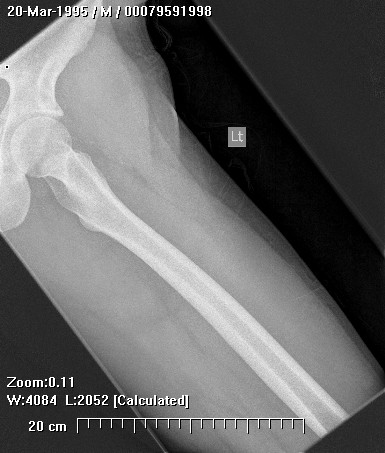
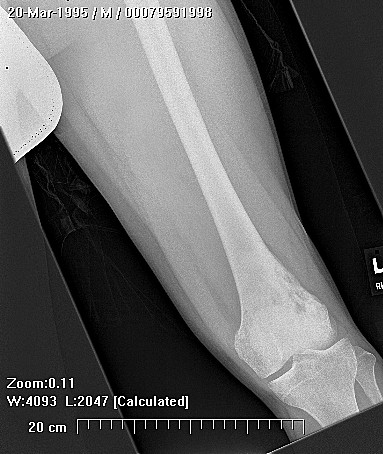
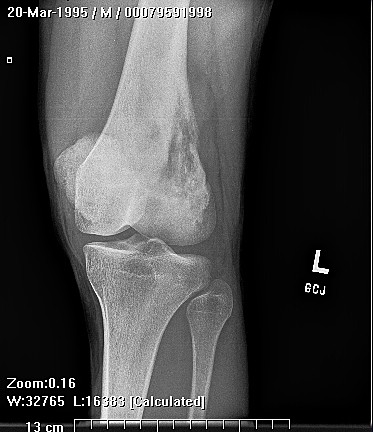
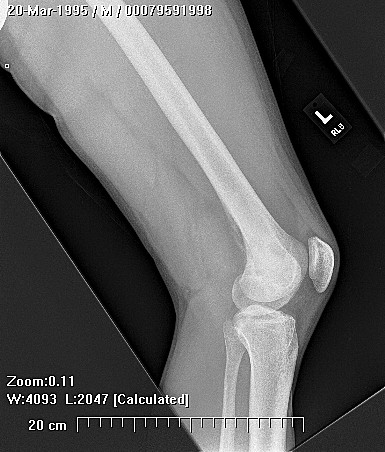
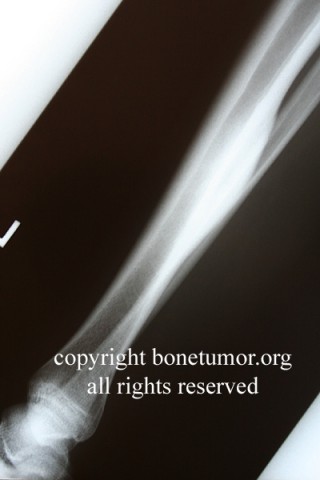
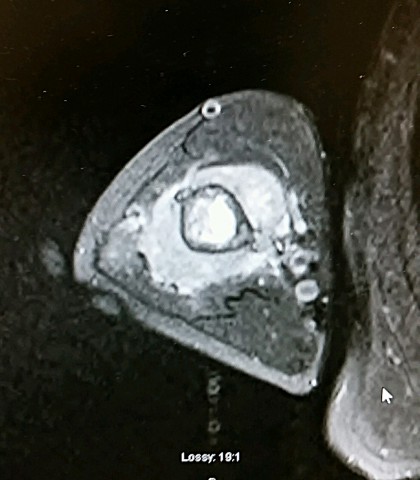
Brief description of the xray
Su apariencia radiográfica muestra márgenes permeantes debido a la presencia del tumor en los canales Haversianos. El sarcoma de Ewing no siempre es obvio en radiografías por lo que otros métodos diagnósticos pueden ser necesarios.
Complete Information on this Tumor
Introduction and Definition
El sarcoma de Ewing es altamente maligno. Es un tipo de tumor periferal primitivo de origen neuroectodermal (PNET). El sarcoma de Ewing se encuentra en las extremidades inferiores más que en las superiores, pero cualquier hueso largo tubular puede ser afectado. La localización más común es en la metafísis y diáfisis del fémur, seguido por la tibia y el humero.
Incidence and Demographics
El sarcoma de Ewing es más común durante la primera y segunda década de vida pero puede afectar a personas de 2 a 80 años de edad. Este tumor afecta más a personas de origen caucásico que a personas afroamericanas y asiáticas. Además afecta mayormente a hombres que a mujeres (razón de 3:2).
Symptoms and Presentation
La presentación clínica del sarcoma de Ewing incluye dolor e hinchazón que dura semanas o meses.
Eritema y calentamiento local pueden ser observados. Típicamente el diagnostico inicial es el de osteomielitis debido a las fiebres intermitentes, leucocitosis, anemia y aumentos en la razón de sedimentación de eritrocitos (ESR).
X-Ray Appearance and Advanced Imaging Findings
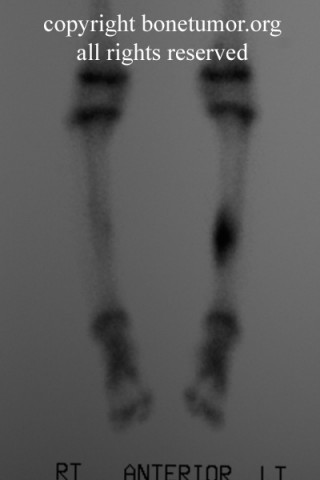
Radiológicamente, el sarcoma de Ewing se asocia con reacciones periosteales lameladas (semejantes a piel de cebolla). Esta apariencia se debe a engrosamientos y divisiones de la corteza ocasionadas por células tumorales. Usualmente, la lesión es lítica y central. Frecuentemente hay desprendimiento endosteal. Durante su progresión la apariencia cambia a un patrón punteado que se extiende al tejido blando. La infiltración de medula ósea no es obvia en radiografías simples. Aunque el sarcoma de Ewing, usualmente es lítico puede presentarse como lesiones escleróticas con expansiones óseas. Estudios de TAC son útiles para definir y cuantificar el grado de destrucción ósea. Estudios de RNM son esenciales para elucidar el envolvimiento de tejidos blandos. En imágenes tipo T1, el tumor presenta baja intensidad comparado a intensidades normales altas de la médula ósea. En imágenes corregidas 1:2 el tumor aparece híper intenso comparado al musculo. El sarcoma de Ewing presenta niveles de absorción aumentadas en estudios de rastreo óseo.
Differential Diagnosis
Infección, metástasis de neuroblastoma, linfoma, leucemia
Preferred Biopsy Technique for this Tumor
Biopsia abierta para lesiones de hueso. Biopsias centrales (de coro) o tru-cut son adecuadas para lesiones en tejidos blandos si el patólogo tiene suficiente experiencia con sarcomas.
Histopathology findings
A nivel macroscópico, el tumor es de colores blanco a grisáceo y pobremente demarcado. La consistencia es blanda y en ocasiones semi-liquida, especialmente luego de rompimientos de corteza. Áreas hemorrágicas y necróticas son comunes. La destrucción ocasionada por la lesión es frecuentemente mayor que la observada por radiografías. Bajo el microscopio, el sarcoma de Ewing consiste de células pequeñas empacadas densa y uniformemente en hojas. Las células tienen citoplasma escaso, un solo núcleo ovalado o redondo ni nucléolos prominentes. Además carecen de bordes distintivos y son de 2-3 veces más grandes que los linfocitos. El tumor se extiende a través de los canales Haversianos lo cual crea una apariencia de márgenes permeantes en radiografías. Las células tiñen positivamente con tintes periódicos de acido Schiff (PAS) debido al contenido de glicógeno. La mayoría de los sarcomas de Ewing tiñen de manera positiva con HBA-71 o 0-13, anticuerpo al producto proteico de myc2.
El diferencial microscópico incluye linfoma y neuroblastoma metastático los cuales deben ser excluidos por medio de tinción con reticulina y análisis en orina de acido vanillil mandélico y acido homovaníllico respectivamente. El rabdomiosarcoma puede ser descartado si la muestra tiñe negativamente con tintes de desmina, mioglobina y actina. Hallazgos de pseudorosetas por microscopio electrónico (EM) apoyan la noción de un posible origen neural. Hallazgos comunes entre el sarcoma de Ewing y tumores primitivos neuroectodermales y la translocación t(11:22)(q24;ql2) también favorecen esta teoría. Se cree que el sarcoma de Ewing con sus pocos organelos, bien podría ser una versión poco diferenciada dentro del espectro de PNET. El neuroepitelioma es un ejemplo de un tumor PNET propiamente diferenciado con gránulos neurosecretorios y procesos neuríticos.
Treatment Options for this Tumor
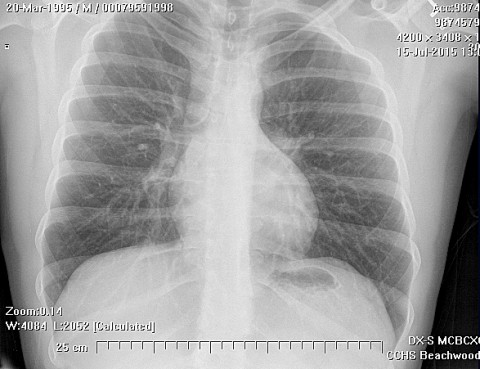
El tratamiento para el sarcoma de Ewing incluye cirugía, radiación y quimioterapia múltiple. La radiación o quimioterapia con Vincristine, dactinomicina y ciclofosfamida (VAC) son utilizadas pre-operativamente. La cirugía es seguida por quimioterapia adyuvante para reducir recurrencias. El tumor se puede metastizar a los pulmones y nódulos linfáticos. Factores que contribuyen a una prognosis pobre incluyen presentación durante edad avanzada, ESR aumentado y leucocitosis.
Preferred Margin for this Tumor
Amplios
Special and Unusual Features
Prognosis pobre en casos de edad avanzada, leucocitosis y valores de ESR elevados.
Suggested Reading and Reference
Eggli, KD et al., Ewing's Sarcoma, Radiologic Clinics of North America, 31(2):325-337, March, 1994.
Bulloughs, Peter, Orthopaedic Pathologv (third edition), Times Mirror International Publishers Limited, London, 1997.
Fletcher, Christopher, Diagnostic Histopathology of Tumors, Churchill Livingstone:New York, 1990.
Huvos, Andrew, Bone Tumors:Diagnosis Treatment and Prognosis, W.B.Saunders, Co., 1991.